










技術 | qPCR不正常曲線分析
溶解曲線主峰左側有峰

一般考慮是引物二聚體或者短的非特異性擴增,解決手段通常有:
◆ 降低引物濃度
20ul體係正常參考引物用量是10uM濃度 0.4ul
◆ 提高退火溫度
梯度摸索引物退火溫度,一般退火溫度不建議超過63°C
◆ 增加模板濃度
當目的基因表達量極低時,染料法QPCR容易形成引物二聚體產物影響實驗結果,提高模板濃度
◆ 重新設計引物
溶解曲線主峰右側有峰
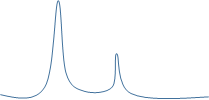
一般考慮非特異性擴增,去除非特異性擴增的手段有:
◆ 提高退火溫度:
梯度摸索引物退火溫度,一般退火溫度不建議超過63°C
◆ 提取的RNA需要去除基因組DNA汙染,或者
◆ 重新設計引物
ROX添加不當
ROX矯正不當可能形成如下熔解曲線圖
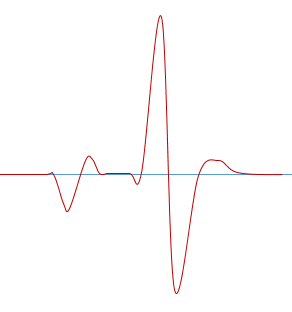
解決手段通常有:
◆ 一定要清楚機器的ROX矯正信息,加的時候一定要看清標簽,高低要分清
◆ ROX要完全融化,混合均勻後再使用
◆ 如果是需要矯正的機器型號但是用了不校正的染料,那麼可以關閉軟件裏麵的ROX矯正選項,重新分析數據
高濃度抑製
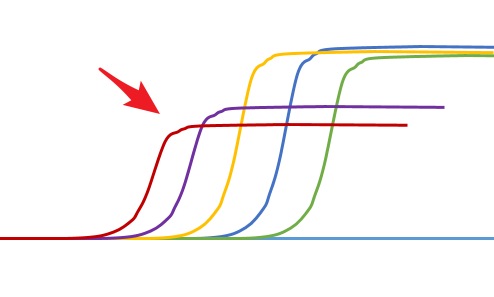
◆ 高濃度模板可能存在一些抑製因素,導致擴增產物的減少,稀釋後抑製就降低或者解除了
◆ 所以如果出現這樣的情況可以調整模板濃度,Ct滯後我們也可以調整下稀釋倍數,保證足夠多的標準點。
濃度梯度Ct值異常

◆ 前麵不能出現梯度,原因可能是模板量過高,超出了線性範圍,那麼需要去掉前麵不能分開的梯度,選擇後麵梯度;
◆ 後麵不能出現梯度,原因可能是模板量低,超出了線性範圍,那麼需要去掉後麵這些梯度,重新摸索高濃度梯度。
無擴增曲線
◆ 機器設定:首先要看下程序的設定方麵,如果程序設定錯誤,那麼是采集不到熒光信號的,
(兩步法擴增程序一般將信號采集設置在退火延伸階段;三步法擴增程序應當將信號采集設置在72°延伸階段)
◆ 循環數:循環數不夠,一般要設置40個循環,但是循環數太多的話會增加背景信號,降低數據的可信度;
◆ 引物降解:可以通過變性PAGE檢測引物是否降解
◆ 引物不合適:重新設計引物
◆ 模板濃度太低: 減少模板稀釋倍數,如果是未知濃度樣品建議從高濃度開始預實驗;
◆ 模板降解:重新製備模板,重複實驗
◆ 表達量低:重新反複設計引物,4-5對不可以放棄該基因
Ct值偏大
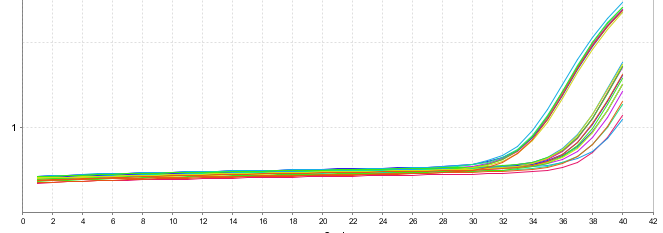
◆ 引物不夠好,擴增效率低,重新設計引物或探針;
◆ 優化PCR條件:
- 改用三步法;
- 增加鎂離子濃度(如果用mix應該也調整不了這個濃度)
- PCR各反應成分的降解或加樣量不足
- 擴增片段太長:建議長度80-200bp
◆ 基因表達量低:
若重新反複設計引物仍不能解決問題,可以放棄該基因或使用其他更為靈敏的檢測方法
擴增曲線先下降後上升

◆ 模板濃度太高,基線的終點值大於CT值:
手動設置減小基線終點值,重新分析數據,(默認的基線範圍一般是3~15個循環,如果遇到擴增在10個循環就起來的那麼收到把基線範圍設置到3~5就可以了,或者是CT值前4~5個循環,基線的設置很可能會影響到引物的擴增效率的結果)
沒有對數增長期
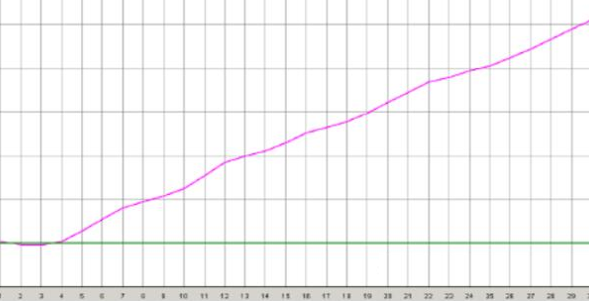
◆ 探針部分降解導致的,如反複凍融探針;在光下暴露時間過長都可能引起,需要重新更換引物探針
◆ PCR反應液中有PCR抑製物,如模板濃度過高,模板中有雜質等
某一空的熒光信號 特別強
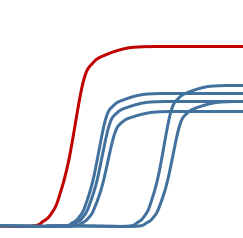
◆ 試劑製備時沒有充分混勻,各管成分不一樣
◆ 如果是探針法的話,可能是探針沒有融化,有一管中的探針量太多
◆ 也可能是機器原因,不過這個可能性不大
擴增曲線有向上的尖峰
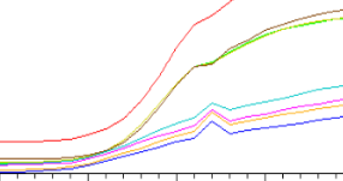
◆ 可能是電壓不穩定導致的,也可能是斷電或者突然開蓋導致的。
山峰形擴增曲線

◆ 反應液蒸發,可能管子沒有蓋好或者管子質量差
標曲標準偏差大於0.5
擴增反應的對數線性期的斜率可用於檢測反應效率。
● 良好的反應效率應在90%-110%之間。
● 擴增效率低於100%,是因為PCR擴增體係中的抑製因素 ,或者RNA有所降解;
● 高於100%是因為非特異性擴增或引物二聚體造成。
解決方案:
◆ 試劑使用前需要徹底融化充分混勻,包括Mix、引物和探針
◆ 使用精確的移液器,加樣時候要垂直加樣

● 擴增信號太弱
● 儀器本身的問題,可能在擴增過程中儀器出現了波動